Ферментная заместительная терапия используется для лечения лизосомных болезней накопления, при которых недостаток ферментов приводит к патологическому накоплению продуктов деградации в лизосомах клеток.
Недостающие ферменты из-за генетических дефектов компенсируются регулярными внутривенными инфузиями. Поскольку введенные синтетические ферменты не могут преодолеть гематоэнцефалический барьер из-за своего молекулярного размера, терапия работает только при лизосомных болезнях накопления, которые не влияют на центральную нервную систему.
Что такое заместительная ферментная терапия?

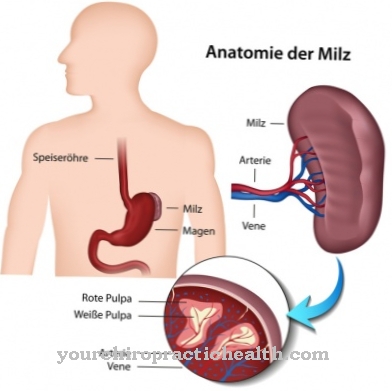
Лизосомы - это особые клеточные органеллы, в которых чужеродные и эндогенные вещества расщепляются и частично рециркулируются. Для разложения и транспорта веществ требуются специальные гидролизующие ферменты. Это протеазы, нуклеазы, липазы и вещества-переносчики.
Ряд известных генетических дефектов может привести к отказу определенных ферментов, так что некоторые продукты деградации накапливаются в лизосомах в патологических количествах и накапливаются, пока не достигнут внеклеточного матрикса, то есть межклеточного пространства, неконтролируемым образом. Все генетические дефекты, которые приводят к неэффективности хотя бы одной необходимой гидролазы, обобщаются под термином лизосомная болезнь накопления. Заместительная ферментная терапия (ERT, заместительная ферментная терапия) используется для замены отсутствующих эндогенных ферментов ферментами, полученными синтетическим путем.
Поскольку гидролазы состоят из относительно больших молекул, они не могут абсорбироваться из кишечника без предварительного расщепления и инактивации, поэтому их можно вводить только внутривенно. Однако размер молекул фермента также предотвращает пересечение гематоэнцефалического барьера, так что терапия может быть эффективной только при лизосомных болезнях накопления, которые не влияют на центральную нервную систему (ЦНС).
Функция, эффект и цели
Известно более 50 различных лизосомальных метаболических нарушений, каждое из которых может быть связано с моногенетическим дефектом. Лизосомные болезни накопления можно разделить на семь различных классов в зависимости от чрезмерно накопленных веществ из-за существующего дефекта ферментов.
Мукополисахаридозы и олигосахаридозы в первую очередь подходят для ФЗТ. Целью ФЗТ всегда является компенсация дефицита определенного фермента с помощью искусственно введенных ферментов, чтобы остановить болезнь или, по крайней мере, облегчить ее течение. Более подробно, замещающие ферменты доступны для следующих лизосомных болезней накопления:
- Болезнь Гоше
- Болезнь Помпе
- Болезнь Фабри
- Синдром Гурлера-Пфаундлера (мукополисахаридоз I)
- Болезнь Хантера (мукополисахаридоз II)
• Синдром Марото-Лами (мукополисахаридоз VI) • Niemann-Pick B
Болезнь Гоше - наиболее распространенная лизосомная болезнь накопления. Это происходит в трех различных вариантах, два из которых также влияют на нервную систему. В ненейропатической форме особенно страдает селезенка, которая значительно увеличивается и приводит к вторичным повреждениям, таким как анемия и повреждение костного мозга. Типичные симптомы - боль в костях и суставах и нарушение кровообращения. Острый невропатический вариант заболевания имеет тяжелое течение и дает мало шансов на выживание после первых двух лет жизни.
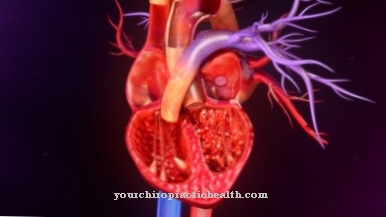
Болезнь накопления Болезнь Помпе возникает из-за дефицита фермента альфа-1,4-глюкозидазы, который участвует в большом количестве метаболических процессов. Болезнь Помпе приводит к огромному увеличению сердца (кардиомегалии) и сердечной недостаточности. Есть ранние, серьезные, курсы, которые появляются в первые несколько месяцев жизни, а также более легкие формы, которые появляются только в более поздние годы жизни.
Болезнь Фабри вызывается генетическим дефектом, сцепленным с Х-хромосомой, поэтому болезнь накопления может поражать только мальчиков и мужчин. Заболевание обычно приводит к появлению симптомов в детском возрасте, включая приступы боли, кератомы кожи, проблемы с почками и повреждение сердечной мышцы. Дефицит фермента альфа-галактозидазы A приводит к накоплению церамида тригексозида, который является причиной появления симптомов, которые также могут влиять на вегетативную нервную систему.
Нередко повреждения приводят к сердечному приступу, инфаркту почки или даже инсульту. Синдром Гурлера-Пфаундлера также известен как мукополисахаридоз I типа и вызывается нарушением метаболизма гликозаминогликанов. Заболевание связано с широким спектром симптомов, включая тяжелые умственные нарушения и серьезные изменения скелета. Течение болезни тяжелое, поэтому средняя продолжительность жизни составляет от 11 до 14 лет. Болезнь Хантера соответствует мукополисахаридозу 2 типа и, как и болезнь Гурлера, вызывается Х-сцепленным дефектом. Заболевание характеризуется течением различной степени тяжести, от раннего детства до легкого течения, которое проявляется только у взрослых мужчин.
Из-за наиболее распространенных сердечных симптомов, таких как дефекты сердечного клапана и проблемы с сердечной мышцей, продолжительность жизни варьируется от нормальной до слегка ограниченной. Синдром Марото-Лами (MPS VI) - это один из мукополисахаридозов, который наследуется по аутосомно-рецессивному типу, поскольку причинный дефект гена находится не на X-хромосоме. Заболевание встречается очень редко, один случай на 455 000 рождений. Известны легкие и тяжелые формы.
Симптомы: увеличение печени и селезенки, синдром запястного канала и изменения сердечных клапанов. Ниманна-Пика B - это липидоз сфингомиелина, который является одним из лизосомных заболеваний накопления и вызывается генетическим дефектом на хромосоме 11. В то время как тип B болезни в основном поражает печень и селезенку, тип A также имеет серьезные нейрональные проблемы.
Здесь вы можете найти свое лекарство
➔ Лекарства от болиРиски, побочные эффекты и опасности
Поскольку многие лизосомные болезни накопления, которые можно лечить с помощью заместительной ферментной терапии, протекают тяжело с соответственно более высоким уровнем смертности, если их не лечить, наибольший риск при ФЗТ заключается в том, что выбранный замещающий фермент не работает или работает слишком слабо.
Другой риск заключается не столько в самой терапии, сколько в том, что основное заболевание распознается слишком поздно, так что ФЗТ может прекратиться во время курса, но уже нанесенный ущерб не может регрессировать. Примерно каждый второй пациент, проходящий лечение, временно реагирует на инфузии такими симптомами, как жар и озноб. Причины этого до конца не изучены. Некоторые пациенты реагируют образованием антител, и известны случаи, когда пациенты реагировали высыпанием и бронхоспазмом.


.jpg)


.jpg)





.jpg)
